用于化学动力学模拟的原子神经网络表示的突破
中国科学技术大学(USTC)姜斌教授带领的团队应用原子神经网络(AtNN)在分子、凝聚相和界面系统的化学动力学模拟方面取得了一系列突破。11月16日,WIREsComputationalMolecularScience发表了对他们作品的评论。
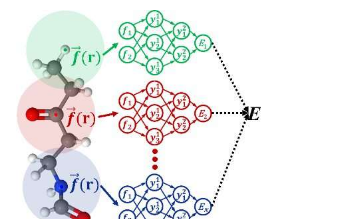
AtNN已广泛用于物理化学研究,特别是化学动力学模拟。AtNN通过将系统属性分解为每个原子的贡献,可以满足分子系统、凝聚相系统和界面系统的不同对称性和周期性,从而实现复杂系统准确高效的分子动力学模拟。
姜斌教授团队发表了一系列关于使用AtNN预测势能面和化学性质的论文。基于他们之前的研究,本文介绍了AtNN方法的基本概念和物理思想,并讨论了在不同的AtNN方法中提高原子环境描述效率的各种策略。
姜斌教授团队提出的嵌入式原子神经网络(EANN)利用高斯型轨道(GTO)线性组合的平方计算三体关联,显着提高了效率。在描述方面,EANN结合了多个径向函数来增强二体相关性。
此外,通过使用消息传递网络递归地合并原子环境之外的非本地交互,与完全基于本地多体描述符的AtNN相比,模型的准确性得到了显着提高。
此外,该论文还总结了最近关于推广标量AtNN模型以表示张量的工作。这些工作主要集中在对AtNN的排列不变输出进行张量化以满足张量的旋转不变对称性——例如,通过坐标和梯度引入方向性,构建一阶或二阶张量来表示响应和过渡偶极矩和极化率等特性。在用AtNN描述电子哈密顿量等更复杂的张量方面也取得了很大进展。
训练数据集对于AtNN中特定系统的模型构建至关重要。江斌教授团队开发的错误搜索等主动学习算法,结合分子动力学轨迹,能够搜索模型中不确定性较大的区域,并采样新的配置加入训练集,从而提高模型训练效果。
数据采样的另一个挑战是有效地考虑原子替换对配置相似性的影响。AtNN在解决上述问题方面的效率通过其在气相表面系统中的几个应用实例得到了证明。
未来,该团队期望将AtNN方法推广到表达长程相互作用、外场下的化学性质,以及求解电子和原子核的薛定谔方程。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。