从人群中挑选出一种细菌
细菌几乎无处不在,对人类和生态健康产生巨大影响。然而,它们对我们来说仍然很神秘。普林斯顿大学MOL教师ZemerGitai、BrittAdamson和NedWingreen发起了一项共同努力,开发新工具来帮助我们更好地了解细菌。他们的工作在《自然微生物学》杂志上发表的一篇论文中进行了描述。
细菌数量惊人,种类繁多。人体肠道中含有的细菌细胞数量与人体全身细胞的数量一样多,分布在大约500-1000种细菌中。细菌也会在我们周围的表面和生物体上繁殖。
同一物种的单个细菌可能表面上相似,但仍然表现出对压力的不同反应,这取决于它们基因的表达模式——也就是说,哪些基因被积极地用来制造最终产品,例如影响细胞行为的蛋白质。
从历史上看,细胞之间的这种类型的变异性很难捕获,因为用于研究细菌的工具只适合对群体范围内的基因表达趋势进行概括。这掩盖了基因表达的差异,而基因表达的差异可以解释抗生素耐药性等重要行为。
最近,科学家们开始利用下一代测序和相关技术,例如单细胞RNA测序,研究人员可以使用它们同时研究数十万个单个细胞的基因表达。然而,仍然存在许多挑战。
例如,其中一些新方法仅限于研究我们已知的一小部分基因,而另一些则受到技术问题的阻碍,从而损害了其敏感性。由研究生布鲁斯·王(BruceWang)带头的普林斯顿大学团队着手克服这些限制。
为了观察哪些基因在细胞中表达,科学家需要寻找一种称为信使RNA(简称mRNA)的分子,这种分子在蛋白质编码基因表达时出现。问题的出现是因为细菌中高达97%的RNA分子是不同类型的RNA,称为核糖体RNA(rRNA)。
大多数方法的工作原理是,首先对细胞中随机选择的所有RNA分子进行标记,然后使用深度测序来发现标记分子的身份。更丰富的rRNA比mRNA更频繁地被测序,导致信噪比较差。Wang和他的同事开发了一种在标记步骤之后、测序步骤之前去除核糖体RNA序列的方法,这既丰富了样品中的mRNA,又降低了测序费用。
王还采用了基于条形码的方法,使这种方法可以同时应用于数十万个细胞。
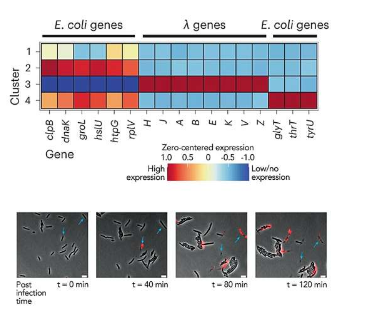
普林斯顿大学的研究小组利用这种方法开始研究细菌生物学的基本问题。例如,他们研究了大肠杆菌对八种不同类型抗生素的反应。
正如预期的那样,不同类型的抗生素会引起不同的反应,具体取决于抗生素的作用方式。然而,各个细胞的反应差异很大,不同的亚群表达不同的基因子集。对相关基因的进一步研究可以帮助科学家开发出针对这些基因的更有效的抗生素或抗生素组合。
研究小组还探讨了细菌如何应对被称为λ噬菌体的病毒感染。令人惊讶的是,他们发现只有三分之一的细菌实际上被λ感染并开始产生其蛋白质,即使噬菌体数量比细菌多100:1。这凸显了只关注整个群体反应的方法可能会掩盖潜在的重要生物复杂性。
这项令人兴奋的新技术被作者称为M3-seq(“大规模并行、多重微生物测序”的缩写),它不仅限于用于单一细菌物种。它还可用于研究多物种细菌群落,例如在人类微生物组中发现的细菌群落,为该团队渴望探索的新领域打开了大门。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。